biodatascience
This is the web resource for NTU's Bio-Data Science and Education Laboratory
Dealing with confounders in omics analysis
There is a theory–practice gap that exists when theoretical statistics are applied on real-world data.
In high-throughput omics data (e.g., genomics, proteomics, and transcriptomics), statistics is commonly deployed to help understand the mechanism underpinning disease onset and progression. In clinical practice, we use statistics to identify differentiating features from high-throughput omics data. These features are critical as biomarkers for diagnosis, guiding treatment, and prognosis. Unlike monogenic disorders, many challenging diseases (e.g., cancer) are polygenic, requiring multigenic signatures to counteract etiology and human variability issues.
In the course of analyzing biological data for differential features such as genes or proteins, we observe that it is a fairly common phenomenon where the null hypothesis is rejected for extraneous reasons (or confounders), rather than because the alternative hypothesis is relevant to the disease phenotype. The situation when null hypothesis are wrongly rejected due to the presence of confounding issues is also known as the Anna Karenina effect [1].
There are many ways a null hypothesis can be rejected. In the example shown, only one is the correct basis for rejection. The rest all lead towards false rejection of the null. The ease with which statistical null hypotheses can be rejected is known as the Anna Karenina effect (so named for the famous addage: Happy families are all alike; every unhappy family is unhappy in its own way).
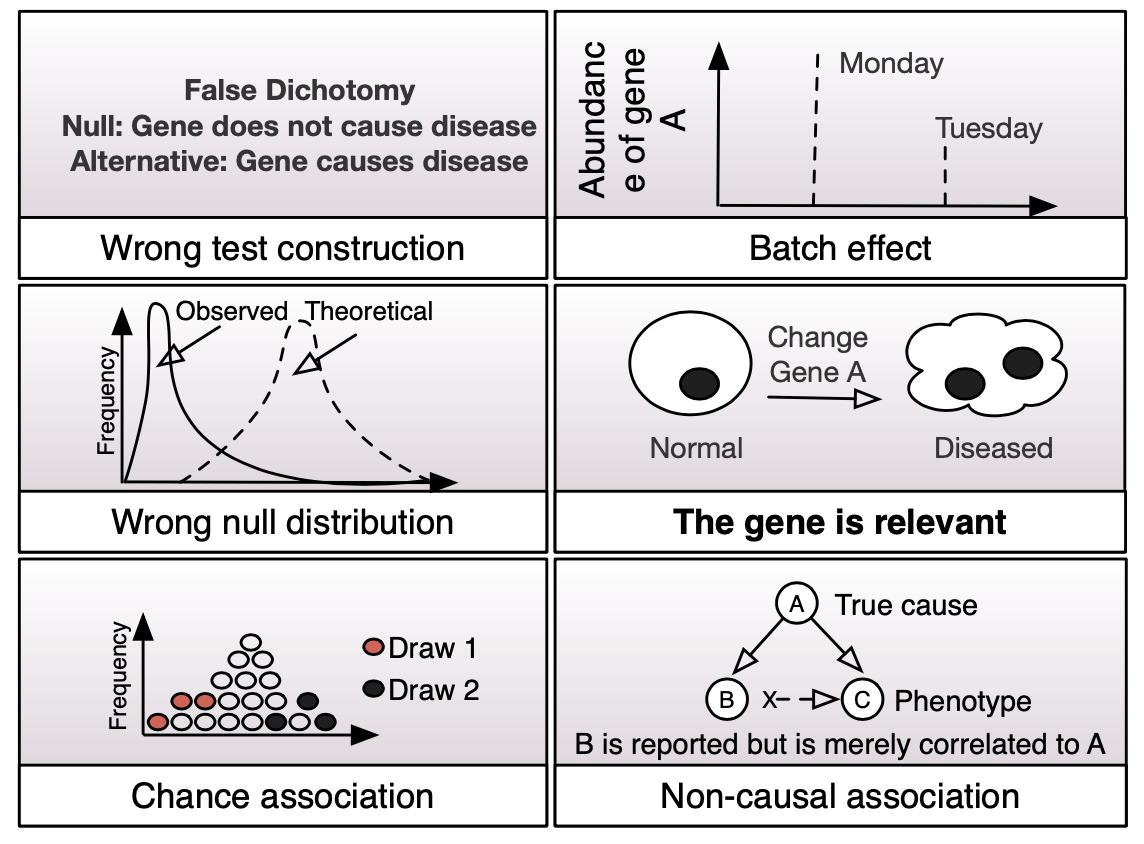
Confounders lead to universality and reproducibility problems. Confounders can include batch effects, poor experiment design, inappropriate sample size, and misapplied statistics. Current literature mostly blames poor experiment design and overreliance on the highly fluctuating P-value. But we think that more can be done.
The subject of this research theme is to explore a deeper rethink on hypothesis statement construction, null distribution appropriateness and relevant test-statistics.
Anna Karenina Effect Checklist
What Causes the Anna Karenina Effect?
- A careless null/alternative hypothesis due to forgotten assumptions:
- Distributions of the feature of interest in the two samples are identical to the two corresponding populations
- Features not of interest are equalized/controlled for in the two samples
- No other explanation for the significance of the test
- Null distribution models the real world
These make it easy to reject the carelessly stated null hypothesis and accept an incorrect alternative hypothesis.
- Good Practices to Avoid Wrong Conclusions and Get Deeper Insight
- Checkforsamplingbias: are the distributions of the feature of interest in the two samples the same as that in the two populations?
- Check for exceptions: are there large subpopulations for which the test outcome is opposite? Are there large subpopulations for which the test outcome becomes much more significant?
- Check the validity of the null distribution: is there evidence that suggests the null distribution is inappropriate?
- Check the hypothesis statement construction: are the hypothesis statements being framed correctly (as opposed to a statement that is prone to being rejected for the wrong reasons)?
- Check your assumptions: are the right assumptions being made (e.g., the independence of measured variables)?
- Check if appropriate summary statistics are used: if an event is extremely rare, then using mean/median-based statistics will miss it; the same goes if many similar events are present, but only one is relevant. Addressing most or all of the above points still does not ensure phenotypic relevance, only correlation.
Reference
[1] Goh WWB, Wong LS. Dealing with confounders in -omics analysis. Trends in Biotechnology, 36(5):488-498, May 2018
Team members
- Wilson Goh
- Limsoon Wong
Relevant publications
- Goh WWB, Wong LS. Dealing with confounders in -omics analysis. Trends in Biotechnology, 36(5):488-498, May 2018
- Goh WWB, Wong LS. Integrating networks and proteomics: moving forward. Trends in Biotechnology, 34(12):951-959, Dec 2016
- Goh WWB, Wong LS. Design principles for clinical network-based proteomics. Drug Discovery Today, 21(7):1130-1138, Jul 2016
- Goh WWB, Wong LS. Computational proteomics: Developing a robust analytical strategy. Drug Discovery Today, 19(3):266-274, Mar 2014
- Zhao Y, Wong LS, Goh WWB. Doing quantile normalization right. BMC Bioinformatics, Accepted
- Goh WWB, Wong LS. Turning straw into gold: how to build robustness into gene signature inference. Drug Discovery Today, 24(1):31-36, Jan 2019
- Goh WWB, Wong LS. Breast Cancer Signatures Are No Better Than Random Signatures Explained. Drug Discovery Today, 23(11):1818-1823, Nov 2018
- Wang W, Sue ACH, Goh WWB. Null-hypothesis statistical testing in clinical proteomics: With great power comes great reproducibility. Drug Discovery Today, 22(6):912-918, June 2017
- Wang W, Goh WWB. Sample-to-sample p-value variability and its implications in multivariate analysis. International Journal of Bioinformatics Research and Applications, 14(3):235-254, Jan 2018