biodatascience
This is the web resource for NTU's Bio-Data Science and Education Laboratory
Batch effects
Batch effects are technical sources of variation, e.g. different processing times or different handlers, which may confound the discovery of real explanatory variables from data.
Batch effects are pervasive and pertinent to all types of high-throughput biological platforms but also to low-throughput methods such as PCR and western blots. Although high-throughput technologies provide higher scalability and coverage, obtaining sufficiently large datasets suitable for clinical analysis requires multiple handlers, reagent batches, machines, segregated running times, etc. Moreover, the burden of data generation is usually borne amongst several collaborating laboratories. In practice, batch effects are almost inevitable.
You might imagine that understanding batches is imperative if we wished to do a good job at big data analysis and also to ensure that whatever gene sets we are evaluating for are truly relevant.
The most common way to deal with batch effects is to attempt to remove them via normalization and/or batch effect-correction algorithms (BECAs). Unfortunately, we overestimate our ability to mitigate batch effects and new problems are emerging: e.g. incomplete removal of batch effects or botched removal attempts given uneven sample-class representation across batches may produce adverse effects.
As today’s data science relies heavily on machine learning and AI tools to learn from weakly-informative variables, ensuring batch effects are mitigated well is even more important than ever: new evidence suggests batch effects specifically inflate cross-validation accuracy — important because cross-validation is the de facto standard for classifier evaluation in the absence of independent validation datasets. Finally and critically, batch effects may obscure/confound biologically important subpopulation effects. Subpopulations are typically under-represented in data and hard to isolate/identify. However, they represent differential etiologies, and so warrant different treatment strategies. Subpopulation effects (and these are clinically valuable) may resemble batch effects, and batch-effect correction can unintentionally remove the former.
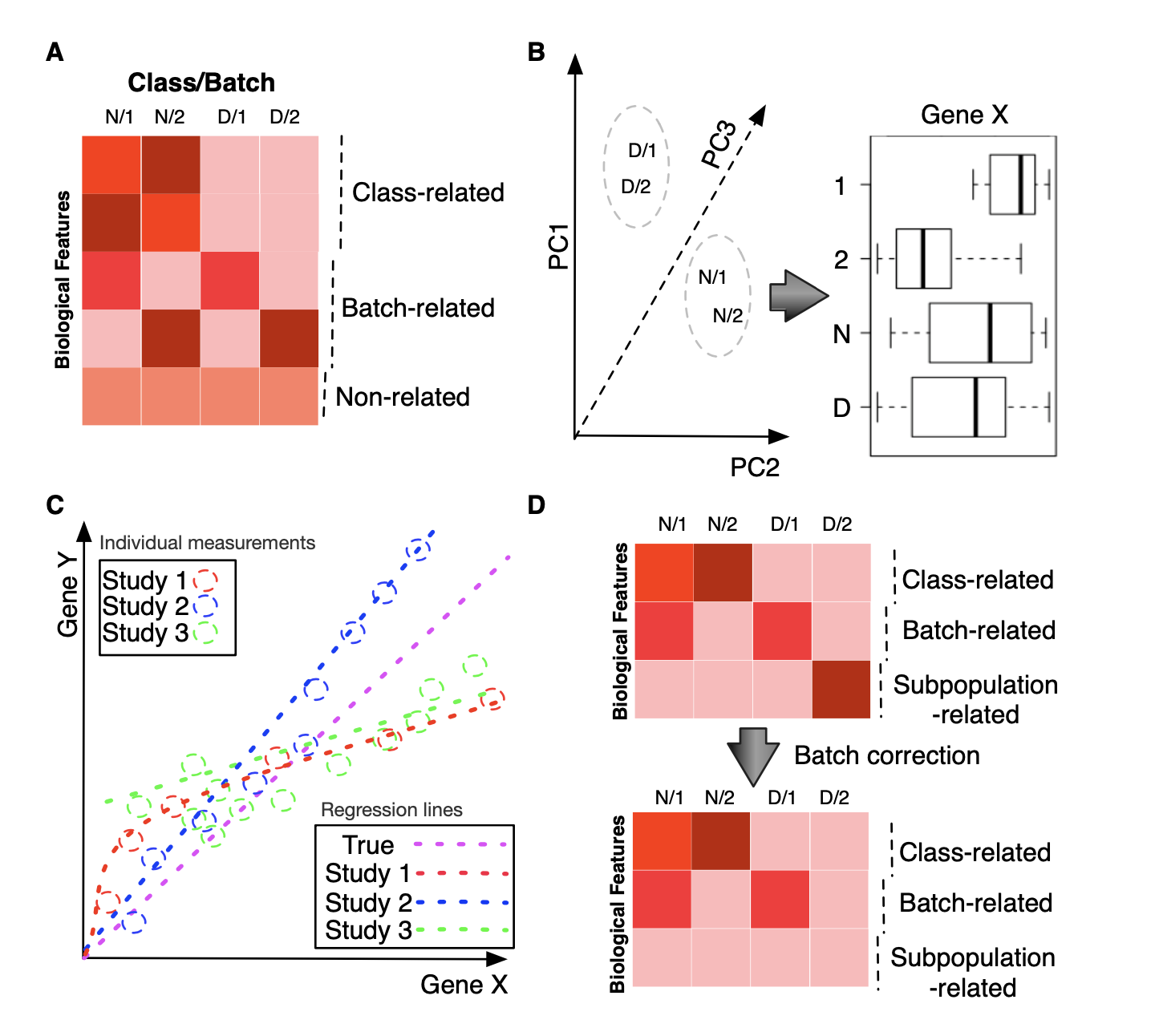
A: We may broadly categorize biological features as those which are associated with class effect (Class-related), batch effect (Batch-related) or completely neutral (Non-related). B: Despite the seeming resolution of batch effect as shown in the principal-component scatterplot (i.e., same class samples aggregate together), individual features may still exhibit strong batch correlation. In this case, a top-ranked feature has stronger correlation with batch (1 and 2) than with class (N and D). C: Meta-analysis does not always agree with each other and can cause us to lose sight of the true picture. Here, we show the correlation between two features, genes X and Y, across studies 1 to 3. The dotted colored lines correspond to the regression for each study, which suggests different relationships between X and Y. The purple dotted line is the true relationship, observable only by combining all three studies. D: Subpopulation effects, if not properly protected, can be lost via batch-effect correction. Information on disease subpopulations can be critical; e.g. in breast cancer, patients are allotted different treatments based on whether they express the estrogen receptor or not. Since certain drugs target the estrogen receptor, patients who do not express the receptor will not benefit from the drug yet have to suffer its potential side effects.
Team members
- Zhao Yaxing
- Zhou Longjian
- Limsoon Wong
- Wilson Wen Bin Goh
Relevant publications
- Zhou LJ , Sue ACH, Goh WWB. Examining the practical limits of batch effect correction algorithms: When should you care about batch effects? Journal of Genetics and Genomics, Accepted (doi.org/10.1016/j.jgg.2019.08.002)
-
- Goh WWB, Wang W, Wong LS. Why batch effects matter in omics data, and how to avoid them. Trends in Biotechnology, S0167-7799(17)30036-7, Mar 2017
- Goh WWB, Wong LS. Protein complex-based analysis is resistant to the obfuscating consequences of batch effects — A case study in clinical proteomics. BMC Genomics, 18(Suppl 2):142, Mar 2017